
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于引力场算法的基因调控网络构建
引用本文
郑明, 刘桂霞, 周柚, 周春光. 基于引力场算法的基因调控网络构建. 2014, 44(2): 427-432[ZHENG Ming, LIU Gui-xia, ZHOU You, ZHOU Chun-guang. Reconstruction of gene regulatory network based on gravitation field algorithm. 吉林大学学报(工学版), 2014, 44(2): 427-432] 
Permissions
Copyright©2014, 吉林大学学报编辑部
版权所有.吉林大学学报编辑部
基于引力场算法的基因调控网络构建
摘要
为了解决传统基因调控网络构建算法准确度不高且效率低下的问题,使用一种基于微分方程的新型网络构建算法。算法分为奇异值分解和引力场算法两部分,奇异值分解策略用来缩小解空间范围,提高运行效率。引力场算法是本文核心,共分初始化、解空间分解、移动算子和吸收算子4步骤。分解策略采用随机分组法,移动算子采用元素逐个移动法,并可根据收敛效果重新移动。最后,将本文算法与另两种启发式搜索算法下的网络构建进行比较,构建模拟和真实的基因调控网络。实验结果显示:本文算法具有更高的执行效率。
关键词:
人工智能; 引力场算法; 基因调控网络; 优化算法; 奇异值分解
中图分类号:TP18
文献标志码:A
文章编号:1671-5497(2014)2-427-6
Reconstruction of gene regulatory network based on gravitation field algorithm
Abstract
In order to resolve the low accuracy and inefficiency of reconstruction of Gene Regulatory Networks (GRNs) in system biology, we proposed a novel inference algorithm from gene expression data based on differential equation model. In this algorithm, two methods are employed for inferring GRNs. One is Singular Value Decomposition (SVD) method and the other one is Gravitation Field Algorithm (GFA). The SVD method is used to decompose gene expression data, determine the algorithm solution space, and get all candidate solutions of GRNs. The GFA is the kernel part of the proposed algorithm. The GFA is divided into four parts: initialization, division of solution space, movement operator and absorption. Random group method is used in division of solution space. Every element movement method is used in movement operator. The proposed algorithm is validated on both the simulated scale-free network and real benchmark gene regulatory network in network database. Both genetic algorithm and simulated annealing are also used to evaluate GFA. The cross-validation results confirm the effectiveness of the proposed algorithm, which outperforms significantly other existing algorithms.
Keyword:
artificial intelligence; gravitation field algorithm; gene regulatory networks; optimal algorithm; singular value decomposition
0 引 言
大规模基因测序技术如mRNA[ 1]杂交微阵列已经极大地增强了研究人员探索基因组层面上生命现象和生命本质的能力。大量数据在数学层面上为各种需复杂计算的问题提供了解决的可能性。用各种基因表达数据进行基因调控网络(Gene regulatory network,GRN)[ 2]构建成为可能,构建算法也成为系统生物学的一个重要组成部分和热点研究领域。GRN的构建主要是指通过一定的数学模型,从基因表达数据或其他形式的数据中推断出基因间相互控制、相互影响、相互促进、相互抑制现象的一系列方法。GRN构建方法多种多样,主要包括布尔网络构建方法[ 3]、贝叶斯网络构建方法[ 4]、神经网络模型构建方法(Neural network model)[ 5]和微分方程构建方法(Differential equation model,DEM)[ 6]等多种形式。各种算法模型所采用的数据是基因表达数据,可通过在线免费数据库如GEO[ 7]中获得。
在众多模型中,DEM是目前已知模型中计算结果最为准确的模型,且可以很好地处理时序数据,但是计算难度相对较大,需要对计算过程进行一系列优化。优化可以从两个方面进行,一个是DEM运算之前采用一定方法确定和缩小DEM的解空间范围,以便于减少后续不必要的运算;另一个则是在DEM运算过程中采用一定优化算法寻找稳定的GRN结构。解空间范围的确定可以采用奇异值分解(Singular value decomposition,SVD)[ 8]方法,但是之前研究往往停留在将解空间以SVD规则求得最终解上。但在RGN构建问题上SVD的最终解却不是最优解,需要继续SVD计算,用以确定GRN构建解空间范围。符合DEM规则的GRN有无限多个,但是最稳定的GRN只有一个或几个,需要采用优化算法在SVD确定的解空间范围内进行优化计算,并最终求得最稳定网络。目前已有的优化算法较多,包括遗传算法(GA)[ 9]、模拟退火算法(SA)[ 10]等,但这些算法不能满足GRN构建问题的优化求解特性。GRN节点在全连接规则下连接数是节点数的平方,所以需要一种能进行大规模计算的新优化算法。
本文采用一种最新的优化算法,即引力场算法(Gravitation field algorithm,GFA)[ 11]来优化和寻找最稳定的GRN。引力场优化算法的特点是运算速度快,同时计算简单,方程更加精确,完全符合GRN构建的优化任务要求。但是由于引力场算法模型本身是连续值优化过程,需要对算法进行改进,以便进行离散问题优化。
1 算法模型
1.1 用SVD确定解空间
DEM可以描述如下:任何一个基因i在时刻t的表达值导数存在如下式所示回归方程特征: 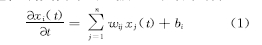
对一个有 N个基因 T个时段的时序基因表达数据,可表示为
X N×T =
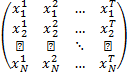 | (2) |
式中: x表示浓度,下标为基因标识,上标为时间序号。
根据式(2),式(1)可写为
d X N×T /d t= W N×N X N×T + B N×T(3)
现将 X N×T进行奇异值分解,得到:
X N×T = U N×T A T×T
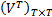 | (4) |
在式(4)中 V与 U都是正交矩阵,而且均满足一定约束条件:
U T U = U -1 U = V -T V = V -1 V = E(5)
在式(5)中 E为单位矩阵,所以 V与 U的逆矩阵等于相应的转置矩阵。为使式(5)成立, U与 V需为广义逆矩阵[ 12]。将式(3)变形并带入式(4)得到:
W N×T =(d X N×T /d t- B N×T) V -T A -1 U -1(6)
式(6)是 GRN的一个特定解,但是此解不一定是 GRN构建所需最优解。需采取一定方法来找出最优解。从式(6)继续出发,可进一步得到算法所需取值范围,即:
W N×T =(d X N×T /d t- B N×T) V -T A -1 U -1 +C U -1(7)式中: C表示任意一个常数。
式(7)是以式(6)作为特解情况下的通解结构。从数学角度讲, W的所有可能解均在公式(7)中。有了此通解后就可在这个缩小的解空间范围内进行 DEM优化求解,并最终找到稳定的 GRN。
1.2 改进引力场算法优化GRN
通过SVD得到DEM通解后,即可在取值范围内进行优化和求解。需要找到一个合理的能量函数,即评价标准来规范DEM。本文采用最小二乘方法来规范这一标准:
c=
 | (8) |
式(8)表示观察值和方程所求值之差的平方,rik是一种残基,具体规定如下:
rik=x'i(t)- 
式中:x'i(t)为t时刻基因i表达值的导数,即变化率:
x'i(t)=xi(t)-xi(t-1)(10)
欲采用GFA求式(8)的最小值,需要由一个权值矩阵组成一个灰尘,矩阵维度与 W相同。这是特殊形式的灰尘格式,但代入 GFA质量函数即式(8)后也应有一个标量结果,作为比较大小的标准。由于矩阵中每个元素的大小的取值范围并不确定,但是绝大多数不会超过 ±100,所以将每个元素的取值范围定为[ -100,100],则灰尘的初始化过程描述如下:
(1)在 N×T次的循环中,每次循环均赋予一个取值在[ -100,100]内的随机值,作为该矩阵灰尘相应位置的元素。
(2)随机初始化完所有 N×T个元素后,将该矩阵代入式(7)中检验是否成立,即是否能求出常数 C值,如能求出,则该灰尘在解空间范围内存在,初始化完毕,否则重新回到步骤(1)再重新初始化。
(3)重复步骤(1)(2)的过程,直到所有灰尘产生。
经过以上3步后完成 GFA的初始化阶段。可见, SVD过程所产生的结果可作为 GFA算法的灰尘取值范围,即相应的解空间,在此基础上可以减少大量不必要的计算,提高 GFA的运行效率。
解空间分解部分采用随机分配方案,因为灰尘的维度过于复杂,平均法不适合解决当前问题。移动算子部分相对特殊,采用如下过程:
(1)对周围灰尘和中心灰尘 N×T个对应元素进行比较,若周围灰尘的元素值不等于中心灰尘相应的元素值,则周围灰尘元素值向中心灰尘元素值靠近,规则采用黄金分割移动形式[ 11]。
(2)进行完 N×T次元素的对比与移动后得到新的灰尘值,将新的灰尘值代入式(7)中进行验证,若满足条件即能求出相应C值,则判定移动成功,进入步骤(4),若不满足条件即求不出相应C值,则进入步骤(3)。
(3)减小移动步伐,使其变为原来的二分之一或三分之一重新进行移动,若已进行P次(P为某一常数)重新移动仍未找到合理移动,则将此周围灰尘删除,即对此周围灰尘直接吸收。
(4)计算与新灰尘值相应的质量函数值,若该值比中心灰尘质量函数值还小即更优,该新值成为新的中心灰尘,原有中心灰尘降为周围灰尘,该组内所有灰尘围绕中心灰尘重新进行移动计算。
经过以上步骤后移动算子结束,吸收算子部分采用标准形式[ 11],即直接删除。至此 GFA在 GRN的应用部分改进结束。在前文所述的权值大小定义中,负值表示抑制作用,即负调控,正值表示促进作用,即正调控,0表示不存在任何调控关系。
在上面所述 GRN构建过程中,计算的是一个全连接的网络,包括节点对自身的控制。但是事实上 GRN往往不是这种密集网络,而是一种稀疏网络。解决这一矛盾的方法较多,但最直接最有效的方法仍然是阈值截取法,即设定某一阈值,比如0 .5,如果当相应权值的绝对值小于0 .5时,即正负调控的影响力小于0 .5时,将此值记为0,即删除相应影响。正负调控影响大于0 .5时,记作影响存在。
通过微分方程模型,采用 SVD缩小和确定解空间范围,运用 GFA优化网络结构,整个过程流程图如图1所示:
 | 图1 GFA进行GRN构建过程流程图Fig.1 The flow chart of GRN with GFA |
2 实验测试与对比
GFA在GRN构建的实验主要分两部分,一个是模拟数据实验,一个是真实数据实验。
2.1 模拟数据实验
; Se= 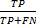
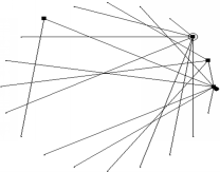
对GRN构建首先要从模拟数据出发,验证网络构建算法的准确性。模拟数据采用算法生成方式获得,首先生成遵从幂律分布的无标度网络[ 13],然后采用DEM的逆向形式获得相应的基因表达数据。根据已有研究,基因调控网络中的控制节点数量很少,且任意一个节点与其他节点连接数都服从幂律分布,一个生成的典型无标度网络结构如图2所示:
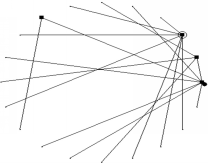 | 图2 典型无标度GRN图Fig.2 A typical scale-free GRN |
在图2中,方块表示GRN调控节点,空心圆表示自调控节点,唯一的实心圆则表示节点0,逆时针顺次为节点1,2,…,19。与此相对应也模拟产生了对应的基因表达数据,部分基因表达数据如表1所示:
| 表1 对应图2产生的部分基因表达数据 Table 1 Part of gene expression data of figure 2 |
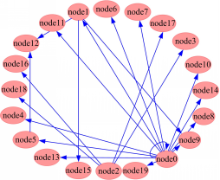
由类似表1所示的基因表达数据,通过图1流程图所示方法进行GRN构建,获得相应的权值矩阵 W,然后通过免费软件 Cytoscape[ 14]进行显示,结果如图3所示:
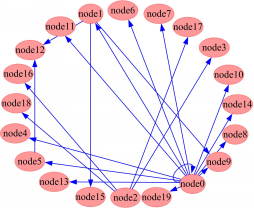 | 图3 基于GFA构建的模拟数据GRN图Fig.3 GRN of simulated data based on GFA |
在图3中箭头表示基因间的相互影响关系,在Cytoscape中采用Circle算法[ 15]进行拓扑布局,所以节点序号发生一定改变,但并不影响算法准确度和运算时间的对比。为比较GFA算法在GRN构建能力方面的优劣,本文使用GA和SA算法进行运算并与本文结果进行对比。
采用Bansal的PPV和Se函数[ 16]进行计算,公式如下:
PPV=
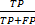 | (11) |
式中: TP=True Positive,表示计算结果为真,实验获得的真实值也为真; FP=False Positive,表示计算结果为假,但通过实验获得的真实值却为真; FN=False Negative,表示计算结果为真,但通过试验获得的真实值却为假。
从式(11)的定义和数学关系来看,PPV和Se都是越大越好,可以根据这两个值来判断算法准确性如何。
除准确度外算法的运算时间也是重要的对比参数。本文将GFA、GA和SA三种算法都用在GRN构建中,且均为图2所示的GRN。运算次数均为500次,将这些不同的结果各自取平均值,总结如表2所示:
| 表2 三种算法分别计算GRN的结果对照表 Table 2 Compared results table of reconstruction of GRN with three algorithms |
从表2中可以看出,GFA算法在PPV和Se上都有一定的优势。运算时间上GFA算法也更快,这是因为SVD方法确定了候选解的解空间范围,加快了运算速度,同时GFA算法本身也可以运算得很快。相比较而言,GFA算法最为出色,但并未超过GA算法太多,SA算法最差,尤其是运算时间过长。
2.2 真实数据实验
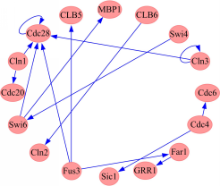
真实数据实验采用的是酵母基因表达数据GDS38,此数据可在GEO数据库获得,对应的GRN结果图可从KEGG数据库获得。本实验只取KEGG酵母GRN图中的一部分基因及其相应的基因表达数据矩阵,采用GFA算法进行GRN优化与构建。此网络子图包含16个基因,如图4所示:
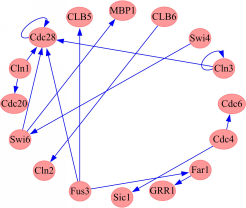 | 图4 基于GFA构建的GDS38部分GRN图Fig.4 GRN of part of GDS38 data based on GFA |
图4是GFA预测的16个酵母基因的结果图,欲得到完整的有统计意义的数据结果,同样需要进行500次GFA、GA和SA的不同实验,结果如表3所示:
| 表3 三种算法分别计算GDS38的GRN的结果 Table 3 Compared results table of reconstruction of GDS38 GRN with three algorithms |
从表3的统计结果可以看出,GFA算法具有更高的准确性,但是与模拟数据相比准确度相对较低,这是因为模拟数据没有其他外界干扰,数据更纯,但不影响GFA与其他算法的比较。同时运算时间也比模拟数据相对要少,这是因为运算节点比模拟数据少了4个。总之,GFA算法在基因调控网络构建过程中可以起到很关键的作用,运算速度更快且运算结果更准。
3 结束语
为提高基因调控网络构建算法的精度并减少算法的运行时间,提出了一种改进的新型优化算法,即引力场优化算法。首先,采用奇异值分解方法获得基因调控网络所有可能的解空间。然后,将整个权值矩阵作为一个灰尘进行初始化。最后,修改移动算子,对每个权值元素都进行移动并检验解空间合法性。将GFA算法利用模拟数据和真实数据分别进行实验,结果表明,与GA算法和SA算法相比,GFA算法的执行效率和计算精度更为出色。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|