{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于高通量数据的人体外源性植物miRNA跨界调控建模
[张浩1, 2  , 占萌苹
, 占萌苹2, 3 , 郭刘香2, 3 , 李誌4 , 刘元宁1, 2 , 张春鹤1, 2 , 常浩武1, 2 , 王志强1, 2 ]
, 占萌苹, 张春鹤|
|


作者简介:张浩(1971-),男,教授,博士.研究方向:生物信息学.E-mail:zhangh@jlu.edu.cn
以基于人体血清的高通量测序数据为研究对象,采用生物计算方法对数据进行分析,以目前已知的人类和植物的RNA数据作为参照集,从中筛选出可能的人体外源植物miRNA;基于miRNA的调控机制原理,获得可能的植物miRNA干扰人类mRNA的靶基因;构建植物miRNA-人类mRNA调控网络模型,分析可能的跨界调控作用。本文通过构建植物-人体跨界调控模型,共挖掘出9个核心模块。研究结果表明,外源性植物miRNA参与调控人体胺代谢、酒精代谢以及脂肪酸代谢等生物过程。
, ZHAN Meng-ping, ZHANG Chun-heExogenous miRNAs are cross-kingdom regulatory in bacteria and viruses, but whether exogenous plant miRNAs are stable in the human body and are involved in cross-kingdom regulation is still controversial. Only by cross-border modeling on the basis of a comprehensive analysis of the human high-throughput miRNA data, and design of effective biological experiments, the human exogenous plant miRNA presence, access pathways and regulatory role can be revealed. In this paper, high-throughput sequencing data based on human serum were used as the study objective and were analyzed by bio-calculation method. The possible human exogenous plant miRNAs were screened by using the RNA data of human and plant as the reference group. Based on the principle of miRNA regulatory mechanism, the possible target genes of plant miRNAs interfering with human mRNA were obtained. A network model of plant miRNA-human mRNA was constructed to analyze possible cross-kingdom regulation. Nine core modules were excavated by constructing a plant-human cross-kingdom regulation model. The results show that exogenous plant miRNAs are involved in the regulation of biological process such as human amine metabolism, alcohol metabolism and fatty acid metabolism.
miRNA是由21~23个核苷酸组成的非编码小分子RNA[1]。它通过与mRNA碱基互补配对结合的方式, 降解靶基因mRNA或者抑制靶基因mRNA的翻译, 对蛋白水平的表达产生影响, 在生物体细胞的生长、分化、凋亡过程中起到重要的调控作用[2]。自miRNA发现以来, 其相关研究主要集中于生物体内源性miRNA的表达和调控机制, 包括miRNA与肿瘤、复杂疾病、流感病毒等的发生和发展关系上, 以及在其他免疫系统之间调控关系的研究[3]。随着研究的深入, 研究人员发现miRNA的调控作用不仅局限于生物体内源, 还可以作为一种信号分子在不同的生命体间进行传导, 对异源细胞的mRNA表达进行调控。现已证实细菌、病毒的miRNA能够在不同宿主间进行传导, 并对宿主mRNA的调控产生影响[2]。真菌及病毒跨界调控的发现为RNA干扰研究领域提出了新的科学问题, 如miRNA的跨界调控是否普遍存在; 如果存在, 外源性miRNA是如何进入宿主体内并参与宿主体内调控; 内源性miRNA与外源性miRNA调控机制有何相关性等。
近期研究还发现, 外源性植物miRNA很可能通过某种途径进入到人体中, 在人体中稳定存在, 并且参与其相关生理调控[4, 5]。Pastrello等[5]实验发现芸苔属蔬菜中含有的miR160致使MTHFD2作为转录物的含量减少; Zhou等[6]发现金银花中的miR2911可以抑制流感病毒的PB1和NS蛋白的合成, 有效地减轻H1N1、H5N1和H7N9亚型流感病毒感染后的症状。Zhang等[7]发现植物miR168a可以与人体的相关靶基因结合, 阻碍LDLRAP1(低密度脂蛋白受体适配蛋白1)的转录, 从而阻碍血浆中低密度脂蛋白LDL的清除。这些研究让科学家们意识到, 外源性植物miRNA对人体也可能存在类似的跨界调控。
由此可见, 外源性植物miRNA可能会对动物产生跨物种调控作用, 但外源性miRNA与内源性miRNA在调控机制上有何关联尚需要进一步研究。目前, 对于在人体检测到的植物miRNA, 是通过饮食等方式进入人体还是由于实验污染所致还未有定论。外源性植物miRNA是否能像内源miRNA一样具有调控作用也需进一步研究。然而, 由于现有可能的外源性植物miRNA均是通过生物实验得到的, 数量少不具有普遍性、且实验结果各异, 严重制约了植物miRNA跨界调控方面的研究。
近年来, 高通量测序技术的迅速发展为解决上述问题提供了数据基础和更好的解决方案。人和动物高通量测序数据的产生、生物计算方法的进步使得全面分析植物miRNA的跨界调控成为可能。
本文通过对收集的miRNA高通量测序数据进行筛选分析, 找出可能存在的外源性植物miRNA, 对其进行靶基因预测, 挖掘出核心模块, 构建植物miRNA-人体miRNA跨界调控模型, 进而研究外源性植物miRNA在人体中可能存在的调控作用。
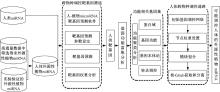
首先, 以NCBI GEO中基于人体血清的高通量测序数据作为研究对象, 采用生物计算方法对数据进行分析, 将目前已知的人类和植物的miRNA数据作为参照集, 从中筛选出可能的人体外源植物miRNA。其次, 基于miRNA的调控机制, 采用分级打分方法预测可能的植物miRNA干扰人类mRNA的靶基因。最后, 构建植物miRNA-人类mRNA调控网络, 分析可能的调控作用。具体流程如图1所示。
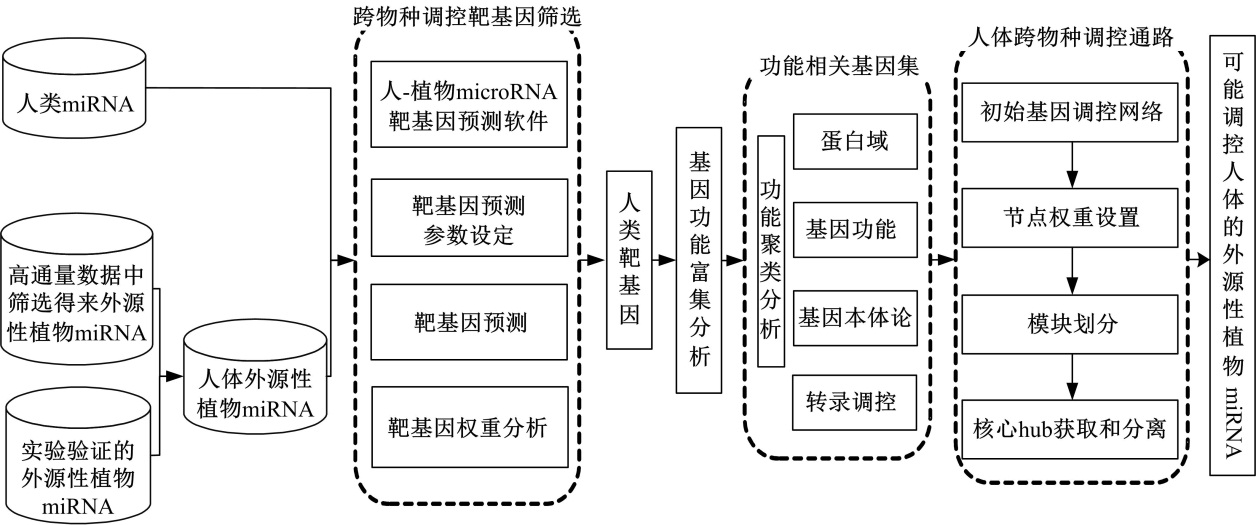 | 图1 外源性植物miRNA功能预测模型Fig.1 Prediction model of exogenous plant miRNA function |
在很多疾病的发生过程中, miRNA在血清、血浆等体液中都异常表达, 参与生物体的调控, 并成为评估身体健康状态的生物标志物。外源性miRNA在人体血清、血浆和其他无细胞样品中被发现, 有可能在参与人体生命活动时发挥重要的调控作用[8]。由于实验过程中检测到的外源性植物miNRA都是在血清组织中, 因此本文采用人体血清组织的高通量测序数据作为研究对象。
本文从NCBI GEO下载人体血浆的高通量小RNA测序数据, 在miRBase数据库下载植物和人的miRNA数据, 在Silva rRNA数据库下载人类的rRNA数据, 在Ensembl数据库下载ncRNA数据, 在Gencode数据库下载mRNA数据。 本文在miRBase下载包括ath(拟南芥), osa(水稻), tae(小麦)和sly(番茄)的71种植物的所有miRNA数据作为标准比对数据集。
(1)高通量数据预处理
由于实验原因, 高通量测序数据中会包含有许多不需要的数据, 如低复杂性的reads和接头会导致错误的组装、PCR扩增中产生的reads重复等。这些片段常出现在长片段插入的文库中, 影响高通量测序数据中成对read (mate-pair)的统计量。因此, 本文需要对数据进行预处理来获取合适的数据。
从测序数据中去除衔接子、测序接头、来自PCR扩增的几乎大量相同reads、制备文库中插入的DNA片段以及poly A的人工污染物。
(2)筛选有效数据
对高通量测序数据而言, 低质量的数据可能存在测序上的错误, 因此需要通过质量分数和k-mer频率修正初步筛选的数据。首先通过脚本将测序数据中低质量分数过滤; 然后过滤出长度小于17 nt和大于26 nt的读数(17和26是miRBase中最短和最长的植物miRNA的长度); 最后得到有效的数据。
(3)挖掘人体外源性植物miRNA
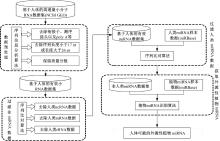
首先, 将获得的合格数据与人体的miRNA、rRNA、tRNA进行blast比对, 在比对过程中允许两个错配, 比对之后将人体的这些小RNA删除; 其次, 将未确认的数据与人类其他RNA(包括ncRNA和mRNA)进行比对, 这次比对中允许一个错配, 并将匹配的数据删除; 最后, 将剩下的数据与植物miRNA比对, 匹配过程中等于或小于一个不匹配, 获得人体所有可能的植物外源miRNA数据。在这个过程中, 使用bowtie软件进行序列比对。植物miRNA筛选流程如图2所示。
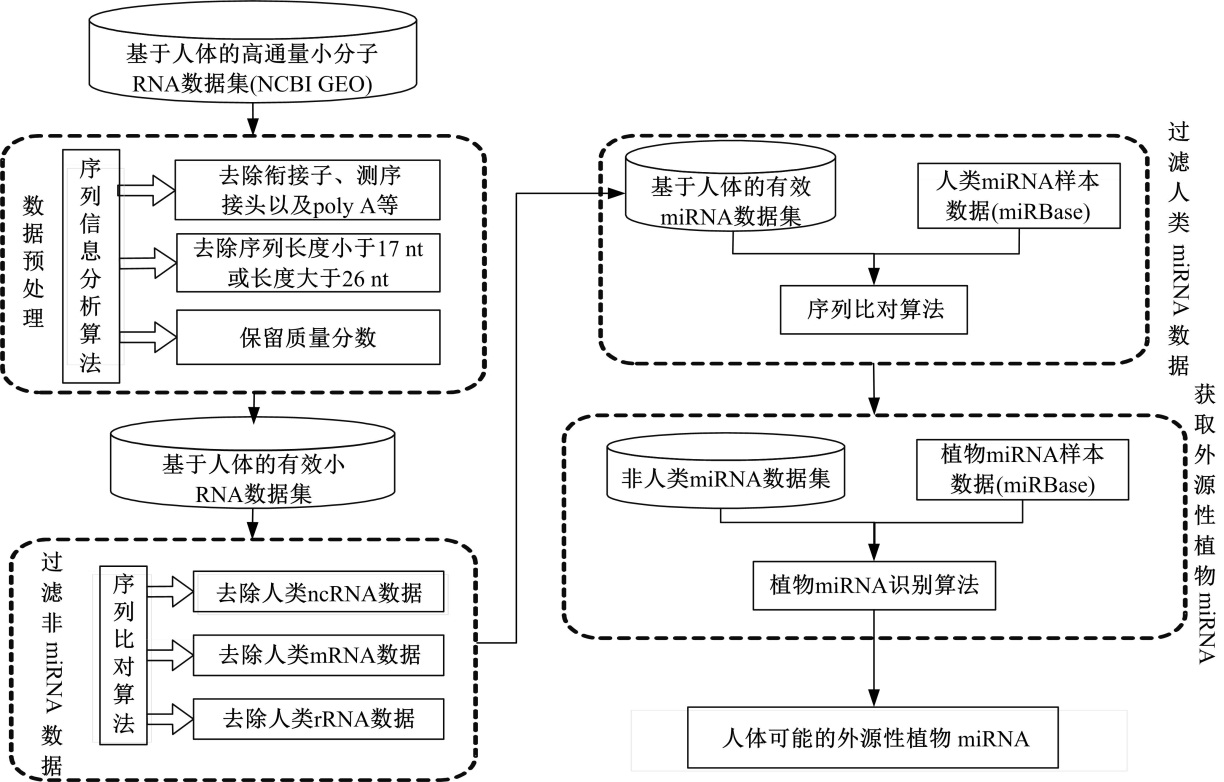 | 图2 基于高通量数据挖掘外源性植物miRNAFig.2 Based on high-throughput data mining exogenous plant miRNA |
本文基于动物体内源性miRNA调控原理, 考虑多靶点, 同时引入结构信息, 确定可能的miRNA-mRNA关联关系。基本规则如下:
(1)将miRNA 5'端的第1~8个核苷酸作为种子区域。
(2)若miRNA与靶标结合的位点位于mRNA的茎区上, 则认为该位点成为miRNA的靶标作用点的可能性大。
(3)基于多步打分的算法miRScore来寻找与植物miRNA对应的人类mRNA靶基因。
miRScore具体算法如下:
(1)针对人体mRNA序列, 基于人体外源植物miRNA共同的起始结合位点打分。
本文采用滑动窗口方法完成此步。滑动窗口大小为8, 步长为1。当miRNA的种子区域与序列完全匹配时, 记录下该起始结合位点。打分规则如下:首先对于同一条序列的同一个位点, 为种子区域能够与从该位点起的8个核苷酸完全相匹配的mRNA加5分; 其次考虑miRNA除种子区域外的其他核苷酸, 当miRNA的核苷酸与mRNA的核苷酸匹配时, 给该组miRNA-mRNA加5分。
(2)基于全部序列的共同二级结构打分;
根据在第(1)步中已经得到的每个miRNA在序列上的起始结合位点, 以miRNA的长度为基准打分, 打分规则如下:如果miRNA与靶mRNA相结合的核苷酸在共同二级结构的茎上, 那么给该组miRNA-mRNA加10分。
(3)计算miRNA-mRNA组分数的平均值, 分数大于平均值的为结果, 其中分数最高的即为最有可能存在调控关系。如果具有相同分数的miRNA多于一个, 那么使用Vienna RNA软件包中的RNAfold, 结合能最低的miRNA-mRNA组为最终结果。
由于靶基因数量较多, 因此核心节点选择的算法尤为重要。在调控网络节点计算权值时常用PageRank算法[9]。其主要思想是在整个网络中, 初始时赋值给各节点相同的值, 采用二维矩阵迭代相乘, 直到网络中全部的节点分值达到稳定。在该算法中, 从一个节点出发, 访问其他节点的概率相等, 即每个节点的随机跳转概率是相同的。然而在调控网络中, 信息少的节点访问信息多的节点的概率小得多。PageRank算法具体公式如下:
由式(1)可知, PageRank 算法中涉及到随机跳转概率c的选取, 在不一样的条件下参数也不具有通用性。通过比较分析, 本文采用LeaderRank算法计算调控网络节点权值。它能够很好地解决上述两个问题。本文通过在网中加了一个背景节点还有该节点与其他全部节点的边来取代PageRank算法中的跳转概率, 得出没有参数并且更简单的一个算法。在LeaderRank 算法中, 通过一个节点的概率与该节点浏览背景节点的概率是一样的, 当一个节点信息量大时, 本算法使其访问背景节点的概率与节点信息量成反比。在开始阶段除了背景节点
直到使用下述公式迭代达到稳定状态:
特别地, 式(2)中邻接矩阵是n+1阶(包含背景节点)。达到稳定状态时把背景节点的分值
LeaderRank算法在用来表示网络中节点重要性等方面有不错的结果[10], 研究发现, LeaderRank相比于PageRank收敛更快; 也可以从网络中更好地发现重要性大的节点, 挖掘出这些有影响力的节点, 同时还有更强的鲁棒性。这些都使LeaderRank 算法应用很广。
现有研究显示, 在基因网络中一个基因通常能够与其周围互相作用的基因形成基因模块, 从而发挥其特定的生物学功能[11]。上述过程中计算了基因调控网络中的节点权值, 并根据LR权值排序各个节点, 将权值较高的选择出来作为核心节点研究, 并挖掘核心网络模块。接下来本文要设计以核心节点为中心的核心调控网络模块挖掘方法, 找到网络中的核心调控模块, 并对挖掘出的模块进行相关分析, 进而探究其参与的调控通路和产生的主要功能。
调控网络是由基因及其之间的相互作用组成。在网络中节点可以是DNA、mRNA、miRNA以及蛋白等活性分子, 边则是这些活性分子间存在的相互作用, 通常具有特定的生物学含义。此外因基因间的作用是相互的, 所以选择无向图。建立网络的关键是找到基因之间的相互关系, 从而组织成基因调控网络。本文以V为节点(基因), E为边(基因间的相互作用), 用G=(V, E)表示二者之间的关系。核心模块的挖掘就是在图G中寻找包含核心基因节点
(1)与核心节点
(2)核心节点
(3)核心节点相邻关系的传递性:如果
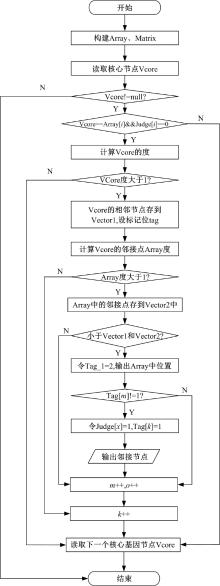
在调控网络中, 挖掘以核心节点为中心的核心网络模块, 具体步骤如下:
(1)导入数据, 包含选取的核心节点、调控网络节点、节点的边对应关系的第1列以及第2列。
(2)创建模型CoreModule类, 在类中以一维数组Array代表节点, 以二维数组Matrix来表示节点以及节点存在联系, 同时定义了初始值为0的一维判定数组Judge, 用来判定核心节点是否在其他核心模块中出现过。
(3)二维矩阵Matrix中根据两节点间是否存在关系进行初始化, 1表示两个节点之间存在关系; 0表示二者不存在关系。
(4)载入核心节点Vcore, 首先判断该核心节点与Array[i](i=1, 2, …, n)是否相等, 同样的Judge[i] (i=1, 2, …, n)是否等于0, 来确认该核心节点是否已经在其他核心模块中出现过。若核心节点与Array[i]相等且Judge[i]等于零, 则计算核心节点的度。若度大于1, 向量Vector1中添加从数组Node中的位置开始的所有与核心节点直接相邻的节点。为了保证A> B和B> A的输出只有一个, 为向量Vector1的值设置了标记, 即定义了一维数组Mark, 其初始值设为0。
(5)设置标记位tag_1, 初始值为0, 对Vcore的每个邻接点Array[x]的处理如下:
(a)计算邻接点Array[x]的度, 如果其度大于1, 那么在向量Vector2中添加Array[x]的直接邻接点。
(b)判断向量Vector1和Vector2中存放的值是否相等, 如果相等, 获取节点在数组Array中的位置y, 把tag_1值设为2(输出标识); 同时Vector1对应的位置为d, 若tag[d]不等于1, 就输出Array[x] > Array[y]节点关系, 并把Judge[x]设为1(表示节点Array[x]已经存在某核心模块中), 并且把tag[k]赋值等于1, 以此来保证A> B和B> A只输出一个。
(c)判断tag_1的值是否等于2, 如果tag_1的值等于2, 则代表邻接点模块已经全部输出, 则需输出核心节点满足Vcore> Array[x]。
(6)重复执行步骤(4)处理完所有核心节点。
本文提取了实验验证的531个过滤靶点的相互作用和信号网络, 然后创建了包含 782个基因和 2444个相互作用的主要调节网络。采用LeaderRank 算法计算调控网络中531个节点的权值, 并且根据权值大小选取出调控网络中的核心节点, 从而挖掘核心模块。对网络中的节点进行计算后, 按照权值大小排序, 部分节点的LR值如下表1所示:
| 表1 节点分值表 Table 1 Node Score Table |
本文选取其中分值最高的前9位(AXIN1, HOXB1, ATP5D, MAPK8IP2, ZAP70, MTNR1B, GPR35, GYS1, C17orf62)作为核心节点进行研究。以下将挖掘以这9个核心节点为中心的核心基因调控网络模块。
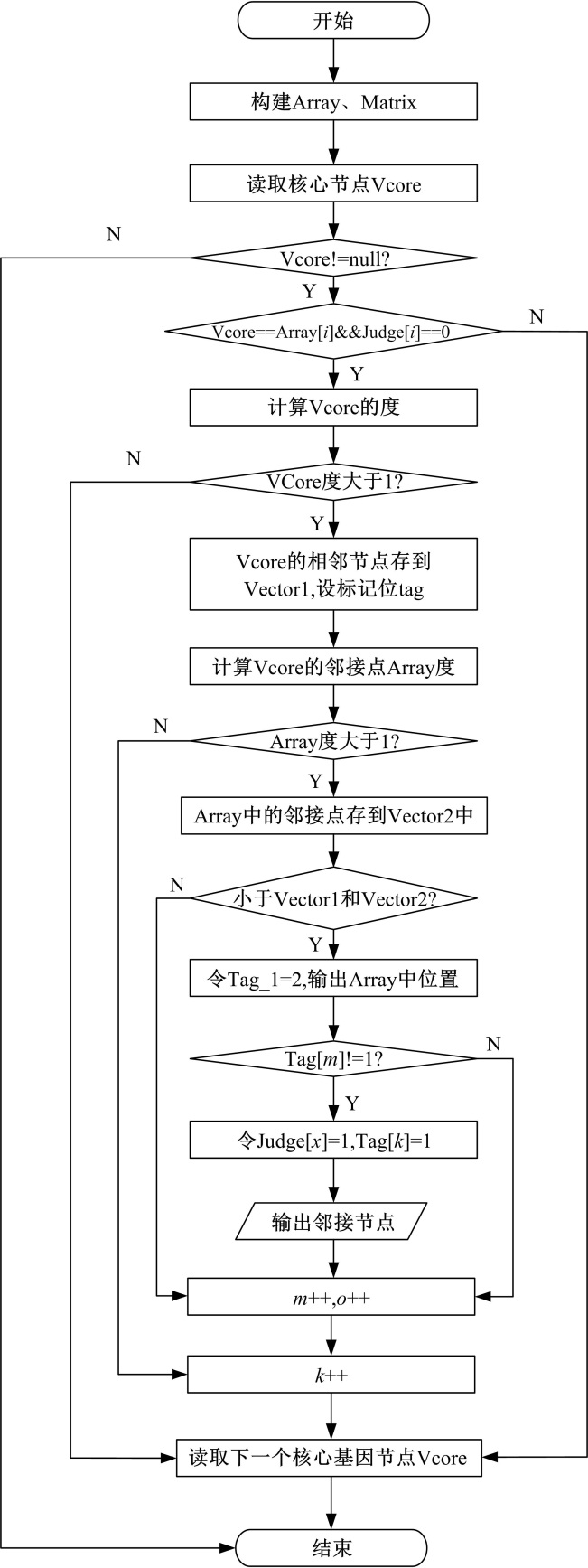 | 图3 核心网络模块挖掘方法流程图Fig.3 Core network module mining method flow chart |
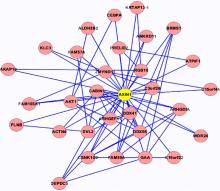
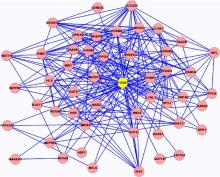
本文在选出核心节点后, 挖掘以核心节点为中心的周围与其相互作用的基因节点形成的核心模块, 然后针对核心基因网络模块进行GO功能富集分析, 探究其在人体中存在的生物学意义。部分核心网络模块如图4和图5所示。
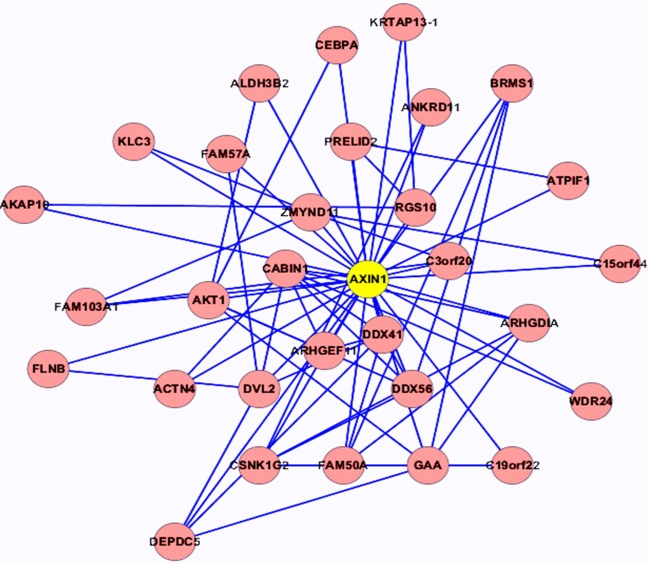 | 图4 AXIN1核心网络模块Fig.4 AXIN1 core network module |
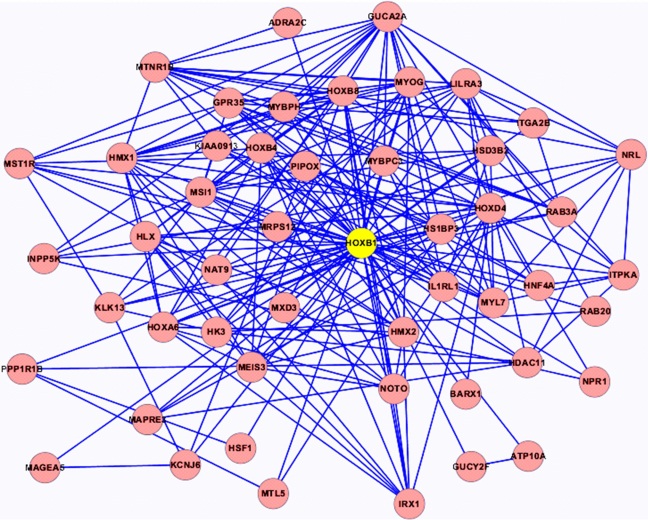 | 图5 HOXB1核心网络模块Fig.5 HOXB1 core network module |
根据功能富集分析的结果, 对各模块进行分析, 部分结果如下:AXIN1涉及核苷三磷酸酶调节活性(GO:0060589)、GTPase激活活性(GO:0005096)、酒精的代谢过程(GO:0006066)、脂肪酸代谢过程(GO:0006631)等; HOXB1涉及胚胎器官发育、铁离子体内平衡、核苷酸的化合物合成过程、芳香族化合物合成过程、RNA代谢过程的调控以及正调控核苷酸化合物代谢过程等; ATP5D涉及免疫系统的调节过程、蛋白氨基酸磷酸化、磷代谢过程、磷酸盐的代谢过程、GYS1涉及嘌呤核苷三磷酸代谢过程、三磷酸核苷代谢过程、氮的化合物的分解代谢过程、ATP的代谢过程; 对这几个核心模块分析发现其多涉及酒精代谢(GO:0006633), 三磷酸核苷代谢(GO:0009141)以及葡萄糖代谢(GO:0006006)等代谢过程。其总体涉及到的部分生物过程如表2所示。
| 表2 核心网络模块功能表 Table 2 Core Network Module Function Table |
由以上的分析结果可以看出, 外源性植物miRNA跨物种调节人类靶基因时, 其靶点多参与人体的日常代谢过程, 这表明饮食习惯的差异, 可能会由于其含有的miRNA不同, 导致摄入人的生命过程受到影响, 人体的代谢过程会发生变化, 导致生理机能的变化。
本文从生物信息学角度, 以从高通量数据中挖掘出的外源性植物miRNA作为出发点, 基于动物miRNA的调控机制, 构建了植物miRNA-人体mRNA跨界调控模型, 研究了外源性植物miRNA在人体中可能存在的调控作用。发现筛选出的植物miRNA与人类的代谢过程关系密切, 并从中挖掘出AXIN1、HOXB1、ATP5D、MAPK8IP2等9个核心节点。由此可见, 饮食习惯可能会由于人体摄入的miRNA的不同导致人体代谢发生变化, 从而导致人体生理机能的变化, 该结果与欧亚人种的饮食和身体机能的关系是相吻合的, 目前还需相关生物实验进行验证。本文提出的跨界调控模型为预测植物miRNA与人体生理及病理相关性提供了可能, 并可以为动物与植物间共进化提供理论依据, 进而从营养学角度揭示日常的饮食习惯对人体生理机能产生的重大影响。同时, 外源性植物miRNA跨界调控研究将为中草药小分子RNA研究找到新的途径, 对我国传统中草药制药具有指导意义, 将有助于探索治疗多种疾病的新思路和新方法。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|