| 1 | KAYSER S, LEVIS M J. Advances in targeted therapy for acute myeloid leukaemia[J]. Br J Haematol, 2018, 180(4): 484-500. |
| 2 | DONG Y, SHI O M, ZENG Q X, et al. Leukemia incidence trends at the global, regional, and national level between 1990 and 2017[J]. Exp Hematol Oncol, 2020, 9: 14. |
| 3 | CREUTZIG U, KUTNY M A, BARR R, et al. Acute myelogenous leukemia in adolescents and young adults[J]. Pediatr Blood Cancer, 2018, 65(9): e27089. |
| 4 | RASCHE M, ZIMMERMANN M, BORSCHEL L, et al. Successes and challenges in the treatment of pediatric acute myeloid leukemia: a retrospective analysis of the AML-BFM trials from 1987 to 2012[J]. Leukemia, 2018, 32(10): 2167-2177. |
| 5 | HU J, LIU Y F, WU C F, et al. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia[J].Proc Natl Acad Sci U S A,2009,106(9):3342-3347. |
| 6 | HARADA K, KONUMA T, MACHIDA S, et al. Risk stratification and prognosticators of acute myeloid leukemia with myelodysplasia-related changes in patients undergoing allogeneic stem cell transplantation: a retrospective study of the adult acute myeloid leukemia working group of the Japan society for hematopoietic cell transplantation[J]. Biol Blood Marrow Transplant, 2019, 25(9): 1730-1743. |
| 7 | WANG Y, HOU J, HE D, et al. The emerging function and mechanism of ceRNAs in cancer[J]. Trends Genet, 2016, 32(4): 211-224. |
| 8 | CHAN J J, TAY Y. Noncoding RNA: RNA regulatory networks in cancer[J]. Int J Mol Sci, 2018, 19(5): E1310. |
| 9 | KARRETH F A, RESCHKE M, RUOCCO A, et al. The BRAF pseudogene functions as a competitive endogenous RNA and induces lymphoma in vivo[J]. Cell, 2015, 161(2): 319-332. |
| 10 | JIANG H, WONG W H. SeqMap: mapping massive amount of oligonucleotides to the genome[J]. Bioinformatics, 2008, 24(20): 2395-2396. |
| 11 | CHOU C H, SHRESTHA S, YANG C D, et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions[J]. Nucleic Acids Res, 2018, 46(D1): D296-D302. |
| 12 | WONG N, WANG X W. miRDB: an online resource for microRNA target prediction and functional annotations[J].Nucleic Acids Res,2015,43(Database issue):D146-D152. |
| 13 | PARK K, KIM K B. miRTar Hunter: a prediction system for identifying human microRNA target sites[J]. Mol Cells, 2013, 35(3): 195-201. |
| 14 | JEGGARI A, MARKS D S, LARSSON E. miRcode: a map of putative microRNA target sites in the long non-coding transcriptome[J]. Bioinformatics,2012,28(15): 2062-2063. |
| 15 | SHANNON P, MARKIEL A, OZIER O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks[J]. Genome Res, 2003, 13(11): 2498-2504. |
| 16 | CASTAIGNE S, PAUTAS C, TERRé C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701):a randomised, open-label, phase 3 study[J]. Lancet, 2012, 379(9825): 1508-1516. |
| 17 | WEI A H, STRICKLAND S A, HOU J Z, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase ib/II study[J]. J Clin Oncol, 2019, 37(15): 1277-1284. |
| 18 | STEIN E M, DINARDO C D, POLLYEA D A, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia[J]. Blood, 2017, 130(6): 722-731. |
| 19 | XIAO H, LIANG S M, WANG L. Competing endogenous RNA regulation in hematologic malignancies[J]. Clin Chim Acta, 2020, 509: 108-116. |
| 20 | SALMENA L, POLISENO L, TAY Y, et al. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language?[J]. Cell, 2011, 146(3): 353-358. |
| 21 | KANG Y Q, ZHANG S P, CAO W J, et al. Knockdown of LncRNA CRNDE suppresses proliferation and P-glycoprotein-mediated multidrug resistance in acute myelocytic leukemia through the Wnt/β-catenin pathway[J]. Biosci Rep, 2020, 40(6): BSR20193450. |
| 22 | YANG Y T, DAI W S, SUN Y W, et al. Long non?coding RNA linc00239 promotes malignant behaviors and chemoresistance against doxorubicin partially via activation of the PI3K/Akt/mTOR pathway in acute myeloid leukaemia cells[J]. Oncol Rep, 2019, 41(4): 2311-2320. |
| 23 | YUAN Z T, WANG W. LncRNA SNHG4 regulates miR-10a/PTEN to inhibit the proliferation of acute myeloid leukemia cells[J]. Hematology, 2020, 25(1): 160-164. |
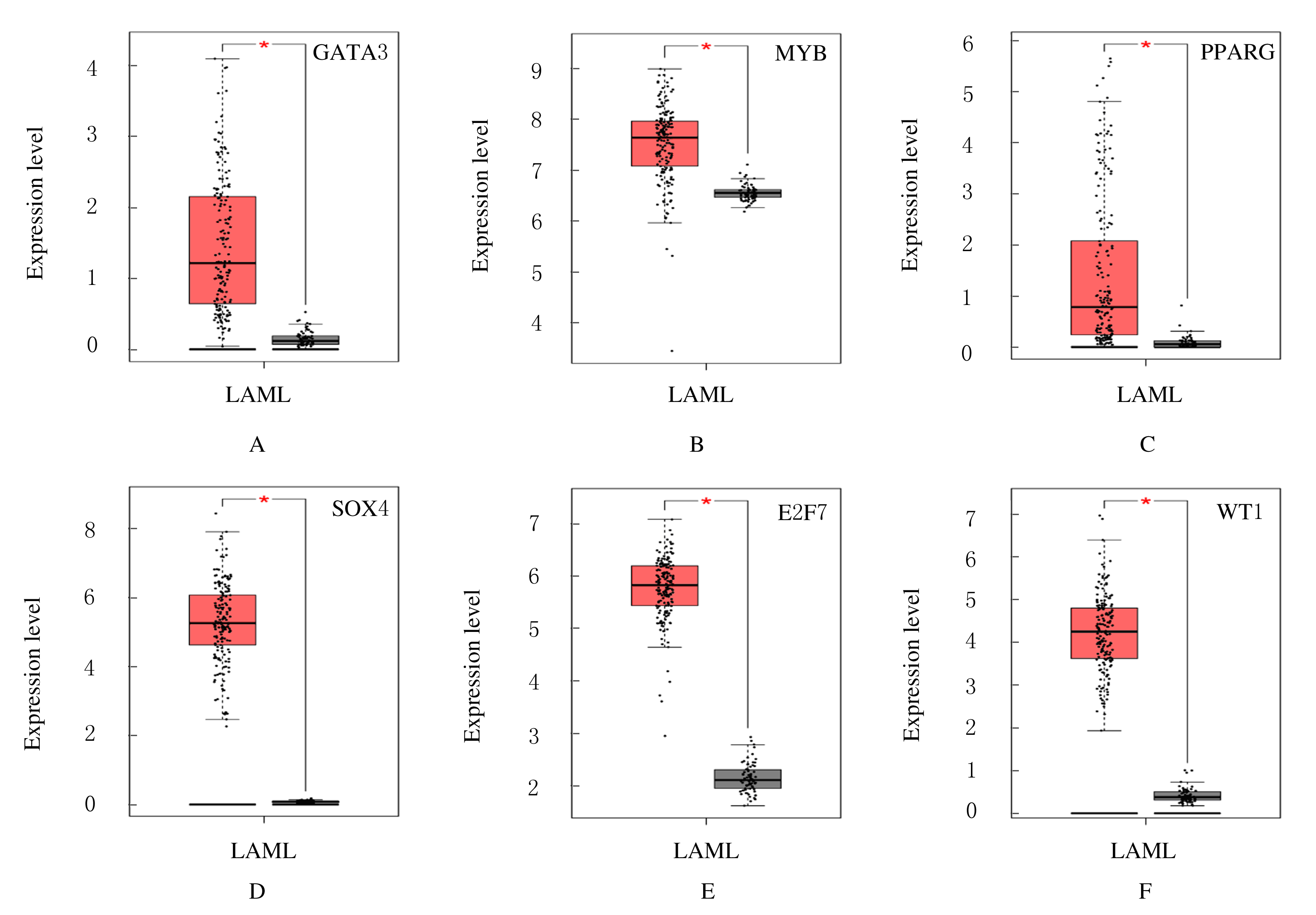
| 24 | FRANSECKY L, NEUMANN M, HEESCH S, et al. Silencing of GATA3 defines a novel stem cell-like subgroup of ETP-ALL[J]. J Hematol Oncol, 2016, 9(1): 95. |
| 25 | YAGUCHI A, ISHIBASHI T, TERADA K, et al. EP300-ZNF384 fusion gene product up-regulates GATA3 gene expression and induces hematopoietic stem cell gene expression signature in B-cell precursor acute lymphoblastic leukemia cells[J]. Int J Hematol, 2017, 106(2): 269-281. |
| 26 | YUSENKO M V, TRENTMANN A, ANDERSSON M K, et al. Monensin, a novel potent MYB inhibitor, suppresses proliferation of acute myeloid leukemia and adenoid cystic carcinoma cells[J]. Cancer Lett, 2020, 479: 61-70. |
| 27 | PAN P, CHEN X. Nuclear receptors as potential therapeutic targets for myeloid leukemia[J]. Cells, 2020, 9(9): E1921. |
| 28 | YING X Y, ZHANG W G, FANG M Y, et al. LncRNA SNHG5 regulates SOX4 expression through competitive binding to miR-489-3p in acute myeloid leukemia[J]. Inflamm Res, 2020, 69(6): 607-618. |
| 29 | ?áLEK C, VYDRA J, CEROVSKá E, et al. WT1 expression in peripheral blood at diagnosis and during the course of early consolidation treatment correlates with survival in patients with intermediate and poor-risk acute myeloid leukemia[J]. Clin Lymphoma Myeloma Leuk, 2020, 20(12): e998-e1009. |
| 30 | SALVATORI B, IOSUE I, MANGIAVACCHI A,et al.The microRNA-26a target E2F7 sustains cell proliferation and inhibits monocytic differentiation of acute myeloid leukemia cells[J]. Cell Death Dis, 2012, 3: e413. |
 ),Yan DU1(
),Yan DU1(