吉林大学学报(医学版) ›› 2020, Vol. 46 ›› Issue (6): 1288-1292.doi: 10.13481/j.1671-587x.20200628
Angelman综合征家系的临床和遗传学特点
程书欢,孙萌,李蒙蒙,程亚颖( )
)
- 河北省人民医院儿科,河北 石家庄 050051
Clinical and genetic characteristics of a family with Angelman syndrome
Shuhuan CHENG,Meng SUN,Mengmeng LI,Yaying CHENG()
- Department of Pediatrics,General Hospital,Hebei Province,Shijiazhuang 050051,China
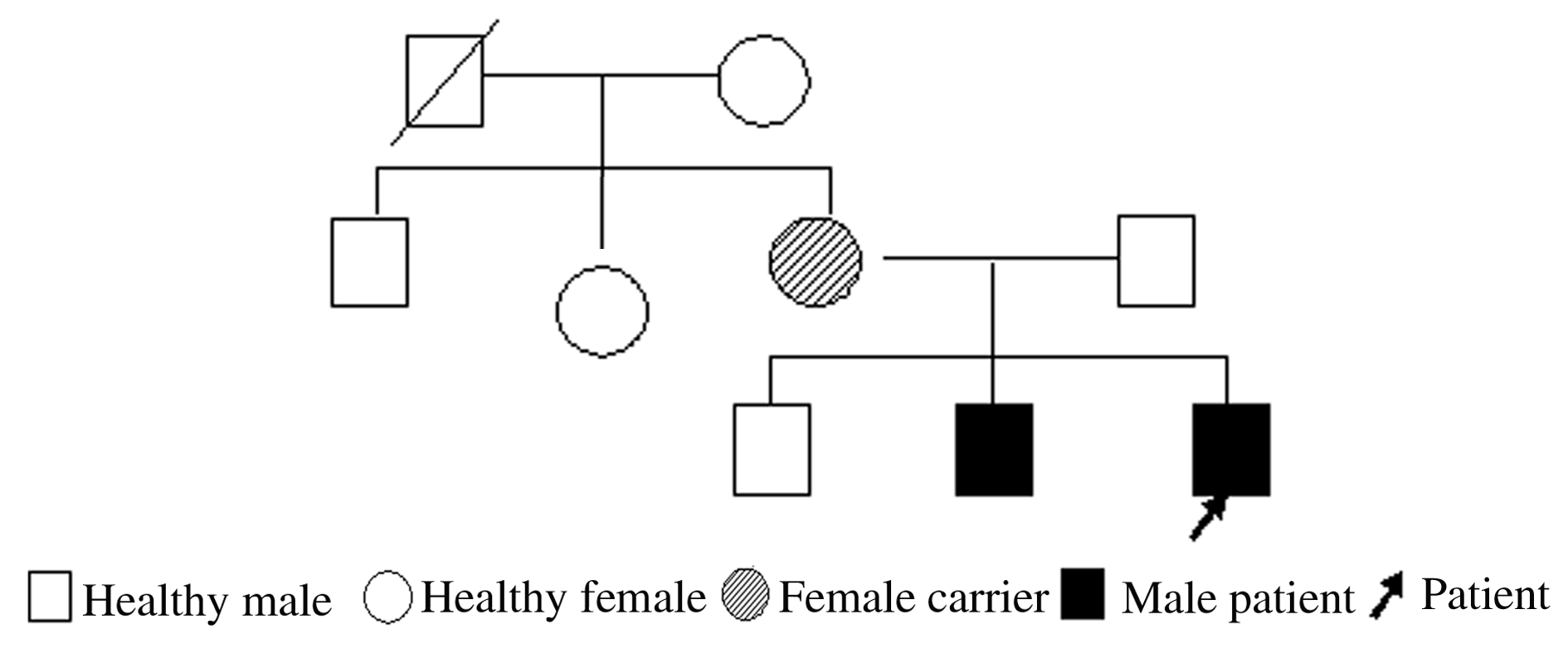
摘要: 总结Angelman综合征(AS)家系的病例资料,分析其临床和遗传学特点,提高临床医生对AS的认识。 收集1个AS家系中两兄弟及其亲属的病史、临床表现、辅助检查和基因检测结果,并进行相关的文献复习。 患儿,男性,年龄4个月3天,表现为发育迟滞、运动障碍、喂养困难;患儿二哥,年龄5岁2个月,表现为严重语言障碍、运动障碍、智力低下、不自觉笑、多动、有异常行为、癫痫发作和特征性脑电图(EEG)等。二代基因测序,患儿及其二哥存在UBE3A基因c.766C>T杂合无义突变,导致氨基酸终止编码。Sanger测序,该突变来源于其母亲。 AS是一种罕见的神经发育障碍性疾病,早期临床表现不典型,需通过分子生物学技术确诊。
中图分类号:
- R729