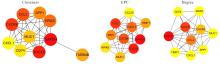
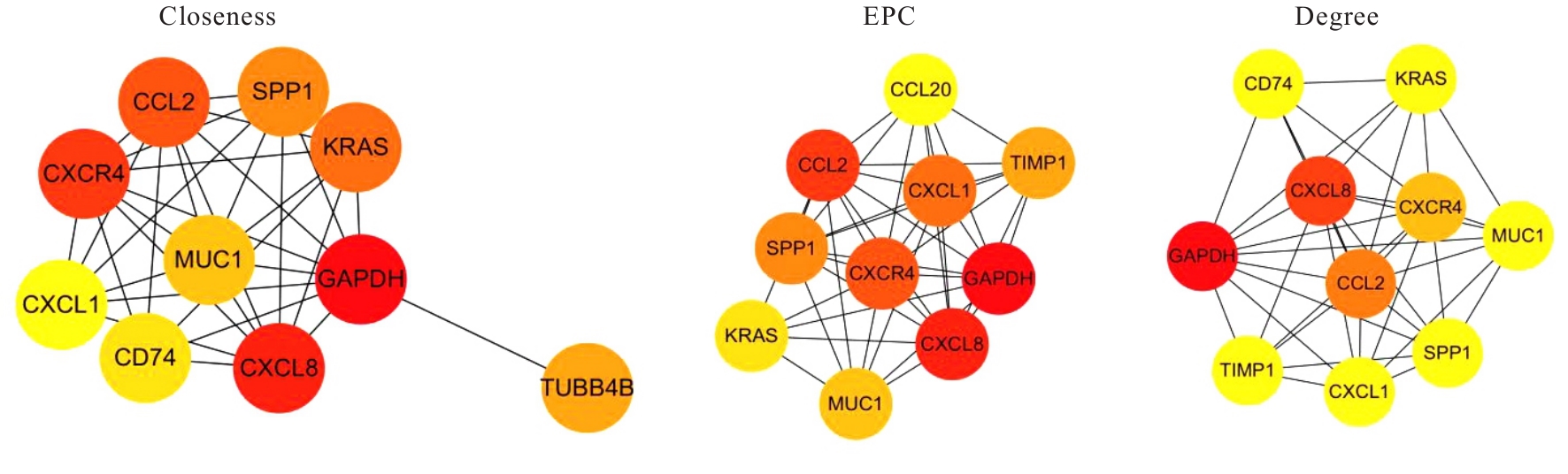
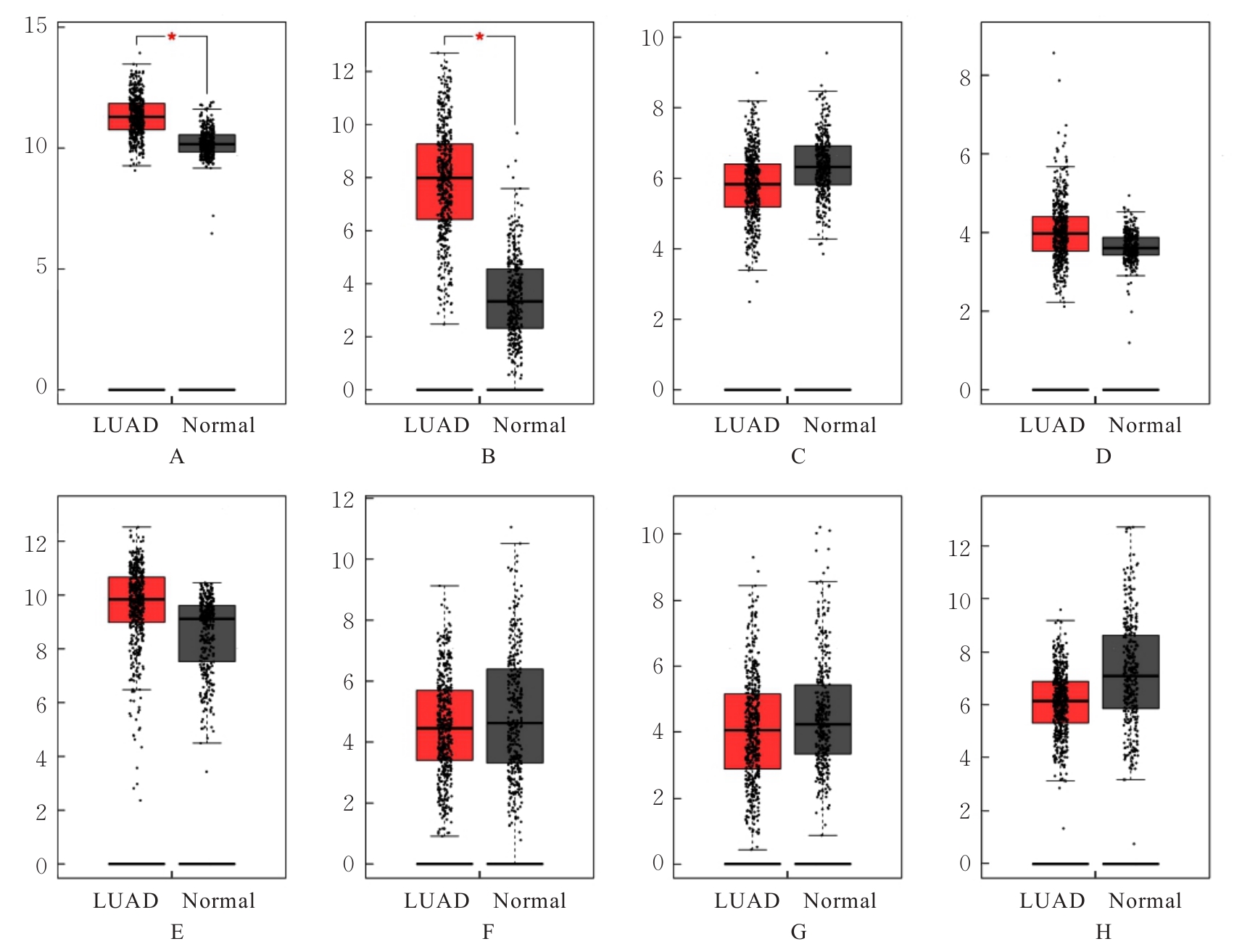
| [1] |
Wenchang CAI,Yuqi LIU,Han WANG,Helin WANG,Zhenjiang WANG,Zishen XIAO,Shiyuan MA,Liping AN,Yanbo LIU.
Expression of protein kinase D2 in bladder cancer tissue and its effect on tumor immune microenvironment
[J]. Journal of Jilin University(Medicine Edition), 2025, 51(2): 378-391.
|
| [2] |
Pengli WU,Fengyu LI,Bo LIU,Yang LYU.
Effect of silencing DDX39A gene on proliferation, migration and invasion of esophageal cancer TE-1 cells and its mechanism
[J]. Journal of Jilin University(Medicine Edition), 2025, 51(1): 115-123.
|
| [3] |
Lyuyin SUN,Zhuping MA,Runlin LI,Yonggang LI,Xiaoli TAO.
Screening of host proteins interacting with Nelson Bay orthoreovirus σNS based on yeast two-hybrid technology
[J]. Journal of Jilin University(Medicine Edition), 2024, 50(5): 1313-1321.
|
| [4] |
Jinlian LI,Lanzhen HUANG,Xishi HUANG,Kangzhi LI,Jiali JIANG,Miaomiao ZHANG,Qunying WU.
Bioinformatics analysis on key genes related to prognosis, diagnosis, and immune cell infiltration of hepatocellular carcinoma and their potential therapeutic drugs
[J]. Journal of Jilin University(Medicine Edition), 2024, 50(4): 1062-1075.
|
| [5] |
Yuanguo WANG,Peng ZHANG.
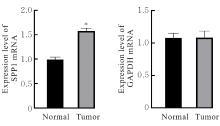
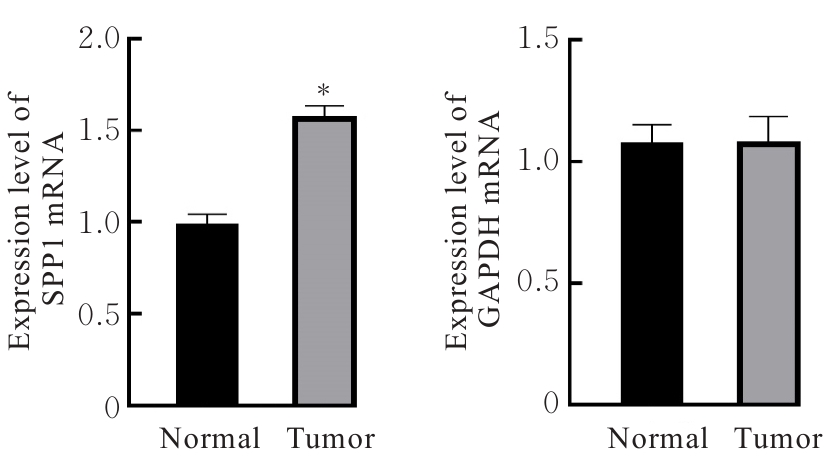
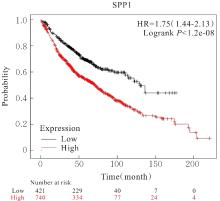
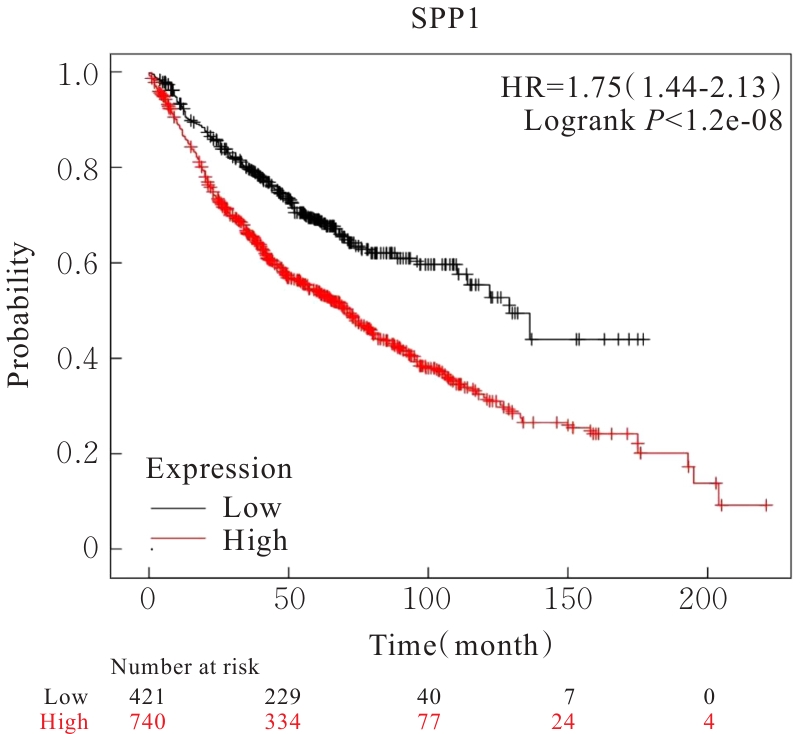
Bioinformatics analysis based on relationship between SSP1 and TGFB1 and occurrence, prognosis, and immune invasion of esophageal adenocarcinoma
[J]. Journal of Jilin University(Medicine Edition), 2024, 50(4): 1076-1086.
|
| [6] |
Linghui LIN,Na LI,Xiaoyan YIN,Xiaoling WANG,Yaping HU,Wei LIU,Rui FEI,Xinli TIAN.
Bioinformatics anlysis based on three-dimensional structure of Helicobacter pylori hp0169 gene
[J]. Journal of Jilin University(Medicine Edition), 2024, 50(3): 739-748.
|
| [7] |
Zijia ZHU,Xia CHEN,Man CUI,Jihong WEN,Ping WANG,Dong SONG.
Bioinformatics and molecular docking technology analysis on mechanism of salidroside on key differential genes of triple negative breast cancer
[J]. Journal of Jilin University(Medicine Edition), 2024, 50(3): 759-769.
|
| [8] |
Yuting LIU,Ying YU,Guizhen LI,Qinxue SHI,Binbin LI.
Bioinformatics analysis based on expression of splicing factor SRSF9 in head and neck squamous cell carcinoma and clinical significance
[J]. Journal of Jilin University(Medicine Edition), 2024, 50(2): 379-391.
|
| [9] |
Liping CHEN,Li HAN,Hua BIAN,Liye PANG.
Bioinformatics analysis based on differentially expressed genes and screening of traditional Chinese medicine for treatment of severe bronchial asthma
[J]. Journal of Jilin University(Medicine Edition), 2024, 50(2): 411-421.
|
| [10] |
Minqi NING,Yong HE,Bingshu LI,Guotao HUANG,Xiaohu ZUO,Zhihan ZHAO,Wuyue HAN,Li HONG.
Bioinformatics analysis based on pelvic organ prolapse related aging genes of GEO Database and LASSO regression algorithm
[J]. Journal of Jilin University(Medicine Edition), 2024, 50(1): 178-187.
|
| [11] |
Zixu YANG,Chang SU,Boyuan WANG,Chong LIU,Minghe LI.
Bioinformatics analysis on molecular subtypes and clinical characteristics of head and neck squamous cell carcinoma based on genes associated with lactate metabolism
[J]. Journal of Jilin University(Medicine Edition), 2024, 50(1): 198-207.
|
| [12] |
Yaqi XU,Yanyu WANG,Wenjing ZHANG,Mei HAN,Huaxia MU,Xi YANG,Weixiao BU,Zikun TAO,Yujia KONG,Fuyan SHI,Suzhen WANG.
Bioinformatics analysis on screening of key genes of hepatitis B virus-related hepatocellular carcinoma and its relationship with prognosis
[J]. Journal of Jilin University(Medicine Edition), 2023, 49(5): 1243-1252.
|
| [13] |
Yiming ZHAO,Haiyang XU.
Bioinformatics analysis on related genes and candidate pathways of glioblastoma multiforme
[J]. Journal of Jilin University(Medicine Edition), 2023, 49(5): 1280-1289.
|
| [14] |
Xiaoying ZHAO,Jiansheng SU,Jiahui MA,Yuefeng WANG,Dan WANG,Liyuan SUN.
Bioinformatics analysis based on activity of heterotrimeric peptide H5LL and construction of recombinant Lactic acid bacteria expression vector
[J]. Journal of Jilin University(Medicine Edition), 2023, 49(5): 1366-1374.
|
| [15] |
Xiaoyan WANG,Yihong HU,Yucheng HAN,Xianqiong ZOU.
Bioinformatics analysis on mechanism of COMMD7 in occurrence and development of brain low-grade glioma
[J]. Journal of Jilin University(Medicine Edition), 2023, 49(2): 414-424.
|
 )
)